Амилоидоз — редкое инфильтративное заболевание, характеризующееся аномальным накоплением амилоида в различных тканях и органах. Амилоид имеет сложное строение и образуется путем сворачивания бета-фибриллярных пептидов, которые откладываются в виде нерастворимого белка в различных местах организма. Амилоидоз1 имеет ряд системных проявлений, в том числе отложение амилоида в головном мозге связано с развитием болезни Альцгеймера и болезни Паркинсона.
Формы амилоидоза
В зависимости от патогенеза различают различные формы системного амилоидоза. Около 90% форм амилоидоза относятся к амилоидозу AL, AA и ATTR.
АЛ-амилоидоз
АЛ-амилоидоз связан с образованием патологического количества легких цепей в результате плазмоцитарного гематологического заболевания, чаще всего миеломной болезни.
MGUS представляет собой моноклональную гаммапатию неопределенной значимости и обычно представляет собой доброкачественное, бессимптомное предраковое состояние, при котором плазматические клетки продуцируют аномальные М-белки, поражающие около 3% людей старше 50 лет. Это состояние требует регулярного наблюдения с интервалом 3–6 месяцев из-за риска прогрессирования в злокачественное новообразование, такое как множественная миелома.
В последние годы заболеваемость АЛ-амилоидозом определенно возросла, и в связи со злокачественным течением заболевания необходимость быстрой диагностики и начала лечения приобретает первостепенное значение.
АА-амилоидоз
АА формы амилоидоза характерны для больных различными хроническими заболеваниями, например, ревматоидным артритом. Его развитие чаще встречается у больных хроническими аутоиммунными или ревматологическими заболеваниями, у которых не достигнут терапевтический контроль воспалительного процесса, что способствует отложению патологического амилоида в тканях.
ATTR транстиретиновый амилоидоз
ATTR транстиретиновый амилоидоз характеризуется наличием различных генетических мутаций, ответственных за неправильное сворачивание фибриллярного белка и образование амилоида. Транстиретиновый амилоидоз, в свою очередь, разделяют на наследственный и ненаследственный (дикий тип), которые имеют различное клиническое течение, возрастную картину и клинический исход.
Наследственный транстиретиновый амилоидоз (hATTR) — редкое прогрессирующее генетическое заболевание, вызванное мутациями гена TTR, приводящее к отложению патологического амилоида в органах. Он вызывает тяжелую необратимую периферическую нейропатию и/или кардиомиопатию, симптомы которой обычно начинаются в зрелом возрасте.
Наследуется по аутосомно-доминантному типу. Мутация гена V122I является наиболее распространенным вариантом Транстиретиновая наследственная сердечная форма амилоидоза.
ATTR дикого типа также называется старческий системный амилоидоз. Этот вариант амилоидоза возникает, когда белок транстиретин вырабатывается печенью в избыточном количестве.
Амилоидоз дикого типа2 чаще поражает мужчин старше 60 лет, характерным для этой формы является поражение сердца.
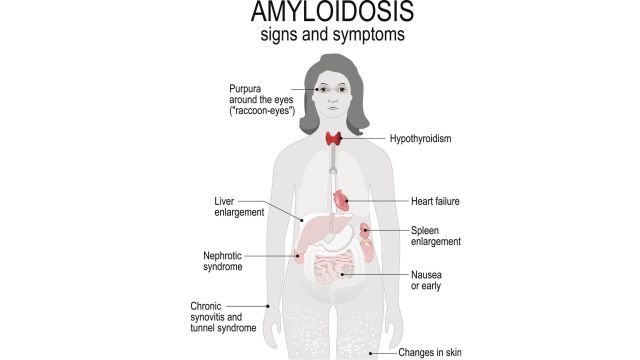
Амилоидоз, протекающий как системное инфильтративное заболевание имеет чрезвычайно разнообразную клиническую картину именно потому, что амилоид можно обнаружить в разных органах.
Клиническая картина амилоидоза
Нередко экстракардиальные жалобы на годы предшествуют кардиальным проявлениям заболевания, при этом причинно-следственная связь между различными жалобами часто упускается из виду. Поражение сердца при амилоидозе происходит с накоплением амилоида в сердечной мышце, клапанах, предсердиях и крупных кровеносных сосудах.
Это приводит к развитию инфильтративная кардиомиопатияпричем пациенты в основном обращаются с жалобами на сердечную недостаточность. В большинстве случаев они сохраняют систолическую функцию левого желудочка, но приводят к появлению симптомов диастолической дисфункции вследствие возникновения ригидности полостей сердца.
Жалобы распространены одышка, утомляемость, снижение физической активности из-за снижения сердечного выброса и болей в груди. Нарушения ритма-проводимости являются частым клиническим проявлением, обусловленным поражением проводящей системы сердца — АВ-блокадой различной степени, фибрилляцией предсердий и желудочковыми аритмиями.
Почечные проявления Для амилоидоза характерен нефротический синдром с иногда массивной протеинурией. Симптомы включают сильное отек ног/лодыжек и чрезмерное количество белка в мочечто делает его пенистым.
Периферическая полинейропатия составляет значительную часть клинической картины больных амилоидозом. Отчеты пациентов онемение, боль или покалывание в руках и ногах.
Вегетативная дисфункция проявляется чаще всего вегетативными жалобами и снижением показателей артериального давления или резким снижением артериального давления после длительной артериальной гипертензии в анамнезе.
Синдром запястного канала и разрыв сухожилия двуглавой мышцы могут быть одними из ранних проявлений заболевания.
Желудочно-кишечные проблемы проявляется гепатомегалией или увеличением печени, болью в животе, необъяснимой потерей веса, диареей, запором и преждевременным насыщением.
Макроглоссия или увеличение языкачто может вызвать затруднения при глотании.
Разнообразие клинических проявлений амилоидоза может помешать своевременной диагностике. Иногда на определение проблемы уходит много лет, и именно обследование и клиническое размышление о возможном заболевании могут привести к постановке диагноза.
Лечение включает терапию уменьшение отложения амилоида в тканях. Лечение транстиретинового амилоидоза предполагает применение двух видов терапии:
- Стабилизаторы амилоида3такие как тафамидис и акорамидис;
- Генные молекулы РНК, такие как вутисиран и патисиран, ингибируют синтез амилоида в печени.
Ссылки:
1. Бустаманте, Жан Г. и Сайед Рафай Х. Заиди. «Амилоидоз». StatPearls, StatPearls Publishing, 2026. PubMed,
2. Дикий тип. Фонд амилоидоза,
3. Верма, Бхупендра и Прити Патель. «Тафамидис». StatPearls, StatPearls Publishing, 2026. PubMed, http://www.ncbi.nlm.nih.gov/books/NBK574508/.